
[ Development of super-resolution imaging technology with less phototoxicity ]
( Tetsuichi Wazawa, Microscopy, 67: 89–98, 2018 )
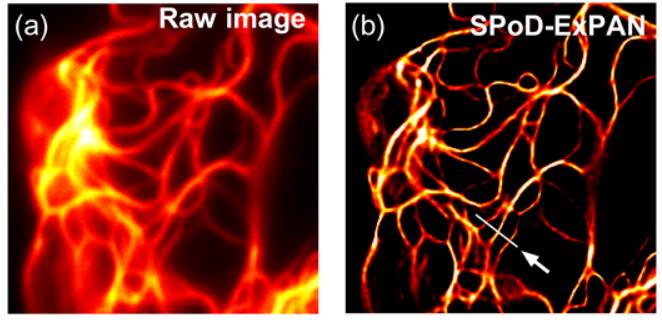
Far-field super-resolution fluorescence microscopy has enabled us to visualize live cells in great detail and with an unprecedented resolution. However, the techniques developed thus far have required high-power illumination (102-106W/cm2), which leads to considerable phototoxicity to live cells and hampers time-lapse observation of the cells. In this study we show a highly biocompatible super-resolution microscopy technique that requires a very low-power illumination. The present technique combines a fast photoswitchable fluorescent protein, Kohinoor, with SPoD-ExPAN (super-resolution by polarization demodulation/excitation polarization angle narrowing). With this technique, we successfully observed Kohinoor-fusion proteins involving vimentin, paxillin, histone and clathrin expressed in HeLa cells at a spatial resolution of 70-80 nm with illumination power densities as low as ~1 W/cm2for both excitation and photoswitching. Furthermore, although the previous SPoD-ExPAN technique used L1-regularized maximum-likelihood calculations to reconstruct super-resolved images, we devised an extension to the Lp-regularization to obtain super-resolved images that more accurately describe objects at the specimen plane. Thus, the present technique would significantly extend the applicability of super-resolution fluorescence microscopy for live-cell imaging.

[ HomoFRET imaging with high-precision polarization microscopy using photonic crystals ]
( Sang-Yeob Kim, Microscopy, 66: 110–119, 2017 )
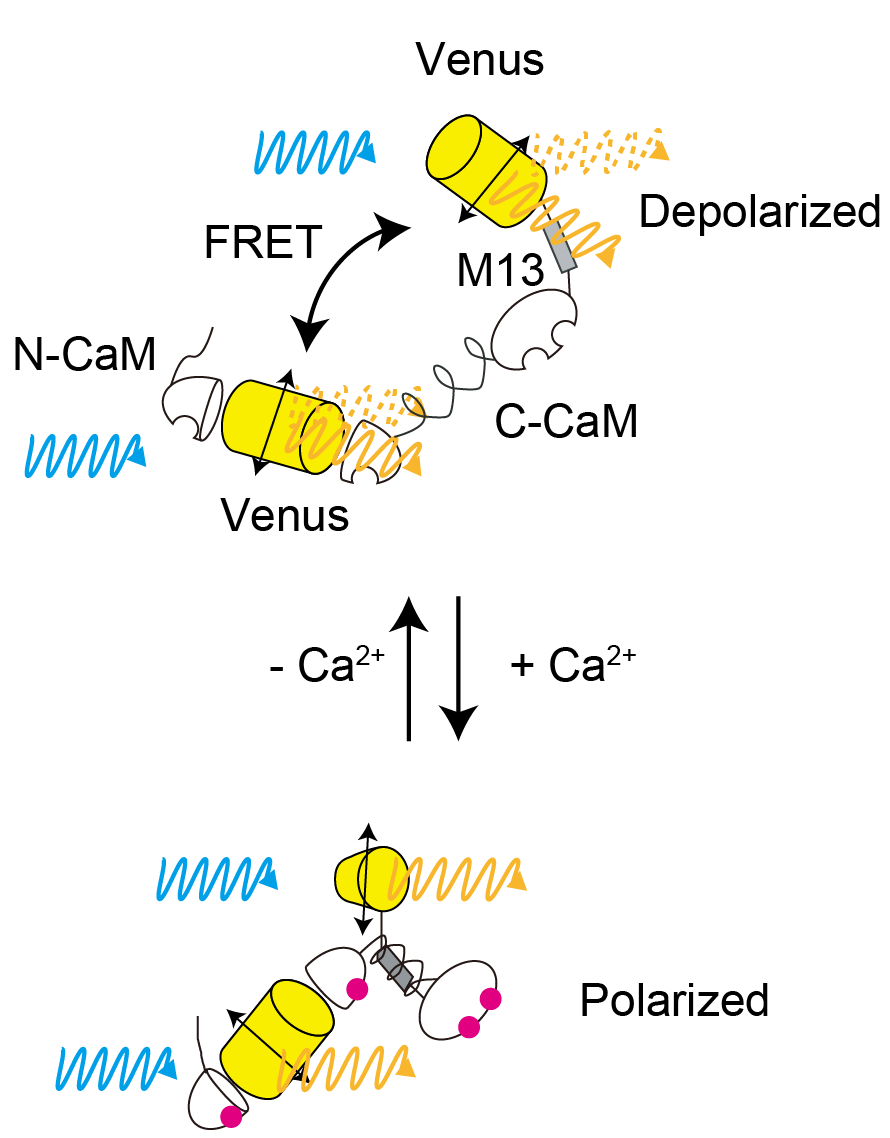
Fluorescent indicators can be used for bioimaging to measure the real time dynamics of intracellular active substances. There are two type of indicators, they are classified as intensiometric types (measuring activity based from the changes of fluorescence intensity) and ratiometric types (measuring activity by using two different indicators). Intensiometric type has one wavelength to measure, it is possible to simultaneously measure multiple fluorescent probes in different wavelengths but the quantitative analysis would be difficult because of the different expression levels of cells. On the other hand, in the "hetero" FRET indicator with two different absorption and fluorescence spectra, by using the ratio value, it is possible to quantitative imaging not depending on the intracellular expression level, it is necessary to measure a wide range of wavelengths, yet It is still difficult to observe multiple intracellular dynamics at the same time. In FRET, it is essential that there is overlap between the absorption spectrum and the fluorescence spectrum, and the same fluorescent probes can be used with a small fluorescent probe with small stokes shift difference. In addition, by using the polarization elimination associated with FRET, it is possible to measure the ratio even though using the same wavelength. This can be done by obtaining the fluorescence anisotropy of the probe. In the observation using dichroic mirror fluorescence microscopy and polarization elimination by coating on the mirror surface, also the polarization elimination effect by using high aperture objective lens still have problem. The problem is the dynamic range of the observed fluorescence anisotropy is small. In order to solve these problems, we developed a "homo" FRET fluorescence indicator using the same fluorescence probe and developed a high-precision fluorescence polarization microscope using photonic crystals. As a Homo FRET indicator, we have developed an indicator w-cameleon that combines with fluorescent protein Venus with Ca2+binding proteins CAM and M13. By using photonic crystals, micro fabrication enables any polarization pattern to be achieved anywhere on the foundation. Therefore, we successfully developed a photonic crystal designed to measure the polarization elimination characteristics of an objective lens and compensate for the elimination of that polarized light. In addition, by eliminating the dichroic mirror by transmission type illumination rather than the incident type, we succeeded in constructing a fluorescence polarization microscope with a high extinction ratio compared with the usage of common linear polarization element.
[ MESSIA ]
( Kenta Saito, Cell Struct. Func., 36: 237-246, 2011 )
Laser-scanning confocal microscopy has been employed for exploring structures at subcellular, cellular and tissue level in three dimensions. To acquire the confocal image, a coherent light source, such as laser, is generally required in conventional single-point scanning microscopy. The illuminating beam must be focused onto a small spot with diffraction-limited size, and this determines the spatial resolution of the microscopy system. In contrast, multipoint scanning confocal microscopy using a Nipkow disk enables the use of an incoherent light source. We previously demonstrated successful application of a 100 W mercury arc lamp as a light source for the Yokogawa confocal scanner unit in which a microlens array was coupled with a Nipkow disk to focus the collimated incident light onto a pinhole (Saito et al, Cell Struct. Funct. 33: 133–141, 2008). However, transmission efficiency of incident light through the pinhole array was low because off-axis light, the major component of the incident light, was blocked by the non-aperture area of the disk. To improve transmission efficiency, we propose an optical system in which off-axis light is able to be transmitted through pinholes surrounding the pinhole located on the optical axis of the collimator lens. This optical system facilitates the use of not only the on-axis but also the off-axis light such that the available incident light is considerably improved. As a result, we apply the proposed system to high-speed confocal and multicolor imaging both with a satisfactory signal-to-noise ratio.
[ CREAM ]
( Yoshiyuki Arai, PLoS ONE, 10:e0125733, 2015 )
The absorption spectrum of light is known to be a "molecular fingerprint" that enables analysis of the molecular type and its amount. It would be useful to measure the absorption spectrum in single cell in order to investigate the cellular status. However, cells are too thin for their absorption spectrum to be measured. In this study, we developed an optical-cavity-enhanced absorption spectroscopic microscopy method for two-dimensional absorption imaging. The light absorption is enhanced by an optical cavity system, which allows the detection of the absorption spectrum with samples having an optical path length as small as 10 μm, at a subcellular spatial resolution. Principal component analysis of various types of cultured mammalian cells indicates absorption-based cellular diversity. Interestingly, this diversity is observed among not only different species but also identical cell types. Furthermore, this microscopy technique allows us to observe frozen sections of tissue samples without any staining and is capable of label-free biopsy. Thus, our microscopy method opens the door for imaging the absorption spectra of biological samples and thereby detecting the individuality of cells.